
Provigil: Léčba bdělosti (úplné informace o předepisování)
Název značky: Provigil
Obecný název: Modafinil
Obsah:
Popis
Farmakologie
Klinické stezky
Indikace a použití
Kontraindikace
Varování
Opatření
Nežádoucí reakce
Zneužívání drog a závislost
Předávkování
Dávkování a správa
Jak dodáván
Informační list pacienta Provigil (modafinil) (v prosté angličtině)
Popis
Provigil (modafinil) je látka zvyšující bdělost pro orální podání. Modafinil je racemická sloučenina. Chemický název pro modafinil je 2 - [(difenylmethyl) sulfinyl] acetamid. Molekulární vzorec je C15H15NO2S a molekulová hmotnost je 273,35.
Chemická struktura je:
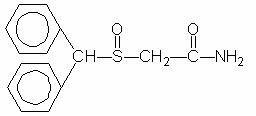
Modafinil je bílý až téměř bílý krystalický prášek, který je prakticky nerozpustný ve vodě a cyklohexanu. Je mírně až mírně rozpustný v methanolu a acetonu. Provigil tablety obsahují 100 mg nebo 200 mg modafinilu a následující neaktivní složky: laktóza, mikrokrystalická celulóza, předželatinovaný škrob, sodná sůl kroskarmelosy, povidon a hořčík stearate.
horní
Klinická farmakologie
Mechanismus účinku a farmakologie
Přesný mechanismus (mechanismy), kterým modafinil podporuje bdělost, není znám. Modafinil má účinky na podporu probuzení podobné sympatomimetickým látkám, jako je amfetamin a methylfenidát, ačkoli farmakologický profil není totožný s profilem sympatomimetických aminů.
Modafinil má slabé až zanedbatelné interakce s receptory norepinefrinu, serotoninu, dopaminu, GABA, adenosinu, histaminu-3, melatoninu a benzodiazepinů. Modafinil také neinhibuje aktivity MAO-B nebo fosfodiesteráz II-V.
Modafinilem indukovaná bdělost může být zmírněna antagonistou ± 1-adrenergních receptorů prazosinem; modafinil je však neaktivní v jiných testovacích systémech in vitro, o nichž je známo, že reagují na ± ± adrenergní agonisty, jako je přípravek potkana vas deferens.
Modafinil není přímo nebo nepřímo působící agonista dopaminového receptoru. Avšak in vitro se modafinil váže na dopaminový transportér a inhibuje zpětné vychytávání dopaminu. Tato aktivita byla spojena in vivo se zvýšenými hladinami extracelulárního dopaminu v některých oblastech mozku zvířat. U myší s genetickým inženýrstvím postrádajících dopaminový transportér (DAT) postrádal modafinil aktivitu na podporu probuzení, což naznačuje, že tato aktivita byla závislá na DAT. Avšak účinky modafinilu na podporu probuzení, na rozdíl od účinků amfetaminu, nebyly antagonizovány dopaminovým receptorovým antagonistou haloperidolem u potkanů. Kromě toho alfa-methyl-p-tyrosin, inhibitor syntézy dopaminu, blokuje působení amfetaminu, ale neblokuje lokomotorickou aktivitu indukovanou modafinilem.
U koček zvyšovaly stejné dávky bdelosti methylfenidátu a amfetaminu zvyšující neuronální aktivaci v mozku. Modafinil v ekvivalentní dávce podporující bdělost selektivně a prominentně zvýšil neuronální aktivaci ve více diskrétních oblastech mozku. Vztah tohoto nálezu u koček k účinkům modafinilu u lidí není znám.
Modafinil produkuje kromě svých účinků podporujících probuzení a schopnosti zvýšit pohybovou aktivitu u zvířat psychoaktivní a euforické účinky, změny nálady, vnímání, myšlení a pocity typické pro jiné stimulanty CNS u lidí. Modafinil má posilující vlastnosti, o čemž svědčí jeho vlastní podání u opic, které byly dříve vyškoleny k vlastnímu podávání kokainu. Modafinil byl také částečně diskriminován jako stimulant.
Optické enantiomery modafinilu mají u zvířat podobné farmakologické účinky. Nezdá se, že by dva hlavní metabolity modafinilu, kyselina modafinil a modafinil sulfon přispívaly k CNS-aktivujícím vlastnostem modafinilu.
Farmakokinetika
Modafinil je racemická sloučenina, jejíž enantiomery mají odlišnou farmakokinetiku (např. Poločas l-isomeru je přibližně trojnásobkem poločasu d-isomeru u dospělých lidí). Enantiomery se navzájem nekonvertují. V ustáleném stavu je celková expozice l-isomeru přibližně třikrát větší než u d-isomeru. Minimální koncentrace (Cminss) cirkulujícího modafinilu po dávkování jednou denně sestává z 90% l-isomeru a 10% d-isomeru. Účinný eliminační poločas modafinilu po opakovaných dávkách je asi 15 hodin. Enantiomery modafinilu vykazují lineární kinetiku po opakovaném dávkování 200-600 mg / den jednou zdravým dobrovolníkům. Zdánlivé ustálené stavy celkového modafinilu a l - (-) - modafinilu jsou dosaženy po 2-4 dnech dávkování.
Vstřebávání
Absorpce tablet Provigilu je rychlá a maximální plazmatické koncentrace se objevují za 2–4 hodiny. Biologická dostupnost tablet Provigil je přibližně stejná jako u vodné suspenze. Absolutní orální biologická dostupnost nebyla stanovena kvůli nerozpustnosti ve vodě (<1 mg / ml) modafinilu, která vylučuje intravenózní podání. Jídlo nemá žádný vliv na celkovou biologickou dostupnost Provigilu; jeho absorpce (tmax) může být zpožděno přibližně o jednu hodinu, pokud je přijato s jídlem.
Rozdělení
Modafinil je dobře distribuován v tělesné tkáni se zdánlivým distribučním objemem (~ 0,9 l / kg) větším než je objem celkové tělesné vody (0,6 l / kg). V lidské plazmě se modafinil in vitro mírně váže na plazmatický protein (~ 60%, hlavně na albumin). Při koncentracích v séru získaných v ustáleném stavu po dávkách 200 mg / den modafinil nevykazuje vytěsnění vazby warfarinu, diazepamu nebo propranololu na proteiny. I při mnohem větších koncentracích (1 000 uM; > 25krát vyšší než Cmax 40 uM v ustáleném stavu při 400 mg / den), modafinil nemá žádný účinek na vazbu warfarinu. Kyselina modafinil v koncentracích> 500 uM snižuje rozsah vazby warfarinu, ale tyto koncentrace jsou> 35krát vyšší než terapeuticky dosažené.
Metabolismus a eliminace
Hlavní cestou eliminace je metabolismus (~ 90%), především v játrech, s následnou renální eliminací metabolitů. Alkalizace moči nemá žádný vliv na eliminaci modafinilu.
K metabolismu dochází prostřednictvím hydrolytické deamidace, oxidace S, hydroxylace aromatického kruhu a konjugace glukuronidů. Méně než 10% podané dávky se vylučuje jako výchozí látka. V klinické studii s použitím radioaktivně značeného modafinilu bylo celkem 81% podané radioaktivity získáno do 11 dnů po podání dávky, převážně v moči (80% vs. 1,0% ve výkalech). Největší zlomek léčiva v moči byla kyselina modafinil, ale nejméně šest dalších metabolitů bylo přítomno v nižších koncentracích. Pouze dva metabolity dosahují znatelných koncentrací v plazmě, tj. Kyselina modafinil a modafinil sulfon. V předklinických modelech byla kyselina modafinil, modafinil sulfon, kyselina 2 - [(difenylmethyl) sulfonyl] octová a 4-hydroxymodafinil neaktivní nebo se nezdálo, že by zprostředkovaly vzrušení účinků modafinilu.
U dospělých se někdy po několika týdnech dávkování pozorovalo snížení minimální hladiny modafinilu autoindukce, ale velikost poklesu a nekonzistence jejich výskytu naznačují, že jejich klinický význam je minimální. Významná akumulace modafinil sulfonu byla pozorována po opakovaných dávkách z důvodu jeho dlouhého eliminačního poločasu 40 hodin. Indukce metabolizujících enzymů, zejména cytochromu P-450 (CYP) 3A4, byla také pozorována in vitro po inkubace primárních kultur lidských hepatocytů s modafinilem a in vivo po prodlouženém podání modafinilu na 400 mg / den. (Další diskuse o účincích modafinilu na enzymatické aktivity CYP viz OPATŘENÍ, Lékové interakce.)
Interakce drog-drogy:
Na základě údajů in vitro je modafinil částečně metabolizován podrodinou izoformy 3A jaterního cytochromu P450 (CYP3A4). Kromě toho má modafinil potenciál inhibovat CYP2C19, potlačovat CYP2C9 a indukovat CYP3A4, CYP2B6 a CYP1A2. Protože modafinil a modafinil sulfon jsou reverzibilní inhibitory enzymu metabolizujícího léčivo CYP2C19, je současné podávání modafinilu s drogy jako diazepam, fenytoin a propranolol, které jsou touto cestou do značné míry vylučovány, mohou zvýšit cirkulující hladiny těchto látek sloučeniny. Navíc u jedinců s nedostatkem enzymu CYP2D6 (tj. 7-10% bělošské populace; podobné nebo nižší v jiných populacích), hladiny substrátů CYP2D6, jako jsou tricyklická antidepresiva a selektivní serotonin inhibitory zpětného vychytávání, které mají pomocné cesty eliminace prostřednictvím CYP2C19, mohou být zvýšeny současným podáváním modafinil. U pacientů léčených těmito a podobnými léky může být nezbytné upravit dávku (viz OPATŘENÍ, Lékové interakce). Studie in vitro prokázala, že armodafinil (jeden z enantiomerů modafinilu) je substrátem P-glykoproteinu.
Současné podávání modafinilu s jinými léčivy působícími na CNS, jako je methylfenidát a dextroamfetamin, významně nezměnilo farmakokinetiku ani jednoho léčiva.
Bylo zjištěno, že chronické podávání modafinilu 400 mg snižuje systémovou expozici dvěma CYP3A4 substráty, ethinylestradiol a triazolam, po perorálním podání, což naznačuje, že byl CYP3A4 indukovaný. Chronické podávání modafinilu může zvýšit vylučování substrátů CYP3A4. U pacientů léčených těmito a podobnými léky může být nezbytné upravit dávku (viz OPATŘENÍ, Lékové interakce).
Po expozici modafinilu in vitro bylo pozorováno zjevné potlačení aktivity CYP2C9 související s koncentrací v lidských hepatocytech což naznačuje, že existuje potenciál pro metabolickou interakci mezi modafinilem a substráty tohoto enzymu (např. S-warfarin, fenytoin). Ve studii interakcí u zdravých dobrovolníků však chronická léčba modafinilem neprokázala významný účinek na farmakokinetiku warfarinu ve srovnání s placebem. (Vidět OPATŘENÍ, Drug Interactions, Other Drugs, Warfarin).
Zvláštní populace
Genderový efekt:
Farmakokinetika modafinilu není ovlivněna pohlavím.
Věkový efekt:
Ve studii s jednorázovou dávkou při 200 mg u 12 byl pozorován mírný pokles (~ 20%) perorální clearance (CL / F) modafinilu. subjekty s průměrným věkem 63 let (rozmezí 53 - 72 let), ale změna nebyla považována za klinicky pravděpodobnou významný. Ve studii s opakovanými dávkami (300 mg / den) u 12 pacientů s průměrným věkem 82 let (rozmezí 67 - 87 let) byl průměr Hladiny modafinilu v plazmě byly přibližně dvojnásobné oproti historicky získaným mladším předmětů. Vzhledem k potenciálním účinkům několika souběžných léků, se kterými byla většina pacientů vzhledem k tomu, že zjevný rozdíl ve farmakokinetice modafinilu nemusí být způsoben pouze účinky stárnutí. Výsledky však naznačují, že u starších pacientů může být clearance modafinilu snížena (viz Dávkování a správa).
Efekt závodu:
Vliv rasy na farmakokinetiku modafinilu nebyl studován.
Renální postižení:
Ve studii s jednorázovou dávkou 200 mg modafinilu nedošlo k závažnému chronickému selhání ledvin (clearance kreatininu 20 ml / min) významně ovlivňují farmakokinetiku modafinilu, ale expozice ke kyselině modafinilu (neaktivní metabolit) byla zvýšena 9krát (viz OPATŘENÍ).
Jaterní postižení:
Farmakokinetika a metabolismus byly vyšetřeny u pacientů s cirhózou jater (6 mužů a 3 ženy). Tři pacienti měli cirhózu ve stadiu B nebo B + (podle dětských kritérií) a 6 pacientů mělo cirhózu ve stadiu C nebo C +. Klinicky 8 z 9 pacientů bylo ikterických a všichni měli ascites. U těchto pacientů byla orální clearance modafinilu snížena asi o 60% a koncentrace v ustáleném stavu byla ve srovnání s normálními pacienty dvojnásobná. Dávka Provigilu by měla být snížena u pacientů se závažným poškozením jater (viz OPATŘENÍ a Dávkování a správa).
horní
Klinické stezky
Účinnost přípravku Provigil při snižování nadměrné ospalosti byla stanovena v následujícím spánku poruchy: narkolepsie, obstrukční spánkový apnoe / hypopnoe syndrom (OSAHS) a porucha spánku (SWSD).
Narkolepsie
Účinnost přípravku Provigil při snižování nadměrné ospalosti (ES) spojené s narkolepsií byla stanovena ve dvou 9 týdnech v USA, multicentrické, placebem kontrolované, dvoudávkové (200 mg denně a 400 mg denně) paralelní skupiny, dvojitě zaslepené studie ambulantních pacientů, kteří splnili Kritéria asociace ICD-9 a American Sleep Diseases Association pro narkolepsii (která jsou také v souladu s American Psychiatric Association) Kritéria DSM-IV). Tato kritéria zahrnují buď 1) opakující se denní spánek nebo přestávky do spánku, ke kterým dochází téměř denně po dobu nejméně tří měsíců plus náhlá oboustranná ztráta posturálního svalového tonu ve spojení s intenzivními emocemi (kataplexie) nebo 2) stížnost na nadměrnou ospalost nebo náhlá svalová slabost s přidruženými funkcemi: paralýza spánku, hypnagogické halucinace, automatické chování, narušený hlavní spánek epizoda; a polysomnografii prokazující jednu z následujících možností: spánková latence kratší než 10 minut nebo spánková latence rychlého pohybu očí (REM) kratší než 20 minut. Kromě toho bylo pro vstup do těchto studií vyžadováno, aby všichni pacienti měli objektivně dokumentovanou nadměrnou denní spavost, vícenásobný spánek Test latence (MSLT) se dvěma nebo více periody REM nástupu spánku a absence jakéhokoli jiného klinicky významného aktivního zdravotnického nebo psychiatrického léčiva porucha. MSLT, objektivní denní polysomnografické hodnocení schopnosti pacienta usnout při nestimulaci prostředí, měří latenci (v minutách) ke spánkovému průměru průměrně během 4 testovacích relací ve dvouhodinových intervalech po nočním polysomnografie. Pro každou testovací relaci bylo řečeno, aby subjekt tiše ležel a pokoušel se spát. Každá testovací relace byla ukončena po 20 minutách, pokud nedošlo k spánku nebo 15 minut po nástupu spánku.
V obou studiích byla primárním měřítkem účinnosti 1) latence spánku, jak bylo hodnoceno testem udržování bdělosti (MWT) a 2) změnu celkového stavu onemocnění pacienta, měřeno pomocí klinického globálního zobrazení změny (CGI-C). Pro úspěšnou zkoušku musela obě opatření prokázat významné zlepšení.
MWT měří latenci (v minutách) ke spánkovému průměru průměrně během 4 testovacích relací ve 2 hodinových intervalech po noční polysomnografii. Pro každou testovací relaci byl subjekt požádán, aby se pokusil zůstat vzhůru, aniž by použil mimořádná opatření. Každá testovací relace byla ukončena po 20 minutách, pokud nedošlo k spánku nebo 10 minut po nástupu spánku. CGI-C je 7-bodová stupnice, se středem beze změny a sahající od velmi hodně horšího po velmi mnoho vylepšená. Pacienti byli hodnoceni hodnotiteli, kteří neměli přístup k žádným jiným údajům o pacientech, než je míra jejich výchozí závažnosti. Hodnotitelé nedostali žádné konkrétní pokyny ohledně kritérií, která měli použít při hodnocení pacientů.
Další hodnocení účinku zahrnovala test vícenásobné spánkové latence (MSLT), stupnice Epworth Sleepiness Scale (ESS; řada otázek určených k posouzení míry ospalosti v každodenních situacích) Steer Clear Performance Test (SCPT; počítačové hodnocení schopnosti pacienta vyhnout se zasažení překážek v simulované jízdní situaci), standardní noční polysomnografie a denní záznam spánku pacienta. Pacienti byli také hodnoceni na stupnici kvality života v narkolepsii (QOLIN), která obsahuje ověřený zdravotní dotazník SF-36.
Obě studie prokázaly zlepšení objektivních a subjektivních měření nadměrné denní spavosti u dávek 200 mg a 400 mg ve srovnání s placebem. Pacienti léčeni jednou dávkou Provigilu vykazovali statisticky významně zvýšenou schopnost zůstat vzhůru na MWT (všechny hodnoty p <0,001) při týdny 3, 6, 9 a závěrečná návštěva ve srovnání s placebem a statisticky významně větší globální zlepšení, hodnoceno na stupnici CGI-C (všechny hodnoty p <0.05).
Průměrné spánkové latence (v minutách) na MWT při základní linii pro 2 kontrolované pokusy jsou uvedeny v tabulce 1 níže, spolu s průměrnou změnou od základní hodnoty na MWT při poslední návštěvě.
Procenta pacientů, kteří vykazovali jakýkoli stupeň zlepšení CGI-C ve dvou klinických studiích, jsou uvedena v tabulce 2 níže.
Podobná statisticky významná zlepšení související s léčbou byla pozorována u dalších ukazatelů poškození v roce 2006 narkolepsie, včetně pacientem hodnocené úrovně denní ospalosti na ESS (p <0,001 pro každou dávku ve srovnání s placebo).
Noční spánek měřený polysomnografií nebyl použitím Provigilu ovlivněn.
Obstrukční spánkový apnoe / syndrom hypopnoe (OSAHS)
Účinnost přípravku Provigil při snižování nadměrné ospalosti spojené s OSAHS byla stanovena ve dvou klinických studiích. Do obou studií byli zařazeni pacienti, kteří splnili mezinárodní klasifikaci poruch spánku (ICSD) kritéria pro OSAHS (která jsou také v souladu s American Psychiatric Association DSM-IV) kritéria). Mezi tato kritéria patří: 1) nadměrná ospalost nebo nespavost plus časté epizody poruch dýchání během spánku a související funkce, jako jsou hlasité chrápání, ranní bolesti hlavy a sucho v ústech probuzení; nebo 2) nadměrná ospalost nebo nespavost a polysomnografie prokazující jednu z následujících situací: více než pět obstrukčních apnoe, z nichž každá trvá déle než 10 sekund, za hodinu spánku a jednoho nebo více z následujících: časté vzrušení ze spánku spojené s apnoe, bradytachykardií a desaturací arteriálního kyslíku ve spojení s apnoe. Kromě toho bylo pro vstup do těchto studií vyžadováno, aby všichni pacienti měli nadměrnou ospalost, jak bylo prokázáno o skóre â ‰ ¥ 10 na stupnici Epworth Sleepiness Scale, a to i přes ošetření trvalým pozitivním tlakem dýchacích cest (CPAP). Spolu s dokumentací o použití CPAP byl vyžadován důkaz, že CPAP byl účinný při snižování epizod apnoe / hypopnoe.
V první studii, 12týdenní multicentrické placebem kontrolované studii, bylo celkem 327 pacientů randomizováno, aby dostávali Provigil 200 mg / den, Provigil 400 mg / den nebo odpovídající placebo. Většina pacientů (80%) byla plně v souladu s CPAP, definovaným jako použití CPAP> 4 hodiny / noc během> 70% nocí. Zbytek byl částečně kompatibilní s CPAP, definovaný jako použití CPAP 30% nocí. V průběhu studie pokračovalo používání CPAP. Primárními měřítky účinnosti byly 1) spánková latence, jak bylo stanoveno testem udržování bdělosti (MWT) a 2) změna celkového stavu onemocnění pacienta, měřeno pomocí klinického globálního zobrazení změny (CGI-C) ve 12. týdnu nebo v závěrečném návštěva. (Vidět Klinické stezky, Popis narkolepsie výše pro popis těchto testů.)
U pacientů léčených Provigilem došlo ke statisticky významnému zlepšení schopnosti zůstávají vzhůru ve srovnání s pacienty léčenými placebem, měřeno pomocí MWT (p <0,001) v koncovém bodě [Stůl 1]. Pacienti léčeni Provigilem také vykazovali statisticky významné zlepšení klinického stavu, jak bylo hodnoceno stupnicí CGI-C (p <0,001) [Tabulka 2]. Obě dávky Provigilu fungovaly podobně.
Ve druhé studii, čtyřtýdenní multicentrické placebem kontrolované studii, bylo 157 pacientů randomizováno buď do skupiny Provigil 400 mg / den nebo do placeba. Dokumentace pravidelného používání CPAP (nejméně 4 hodiny / noc po 70% nocí) byla vyžadována u všech pacientů. Primárním výsledkovým měřítkem byla změna oproti základní hodnotě ESS ve 4. týdnu nebo závěrečná návštěva. Základní skóre ESS pro skupiny Provigil a placebo bylo 14,2, respektive 14,4. Ve 4. týdnu byla ESS snížena o 4,6 ve skupině s Provigilem ao 2,0 ve skupině s placebem, což byl statisticky významný rozdíl (p <0,0001).
Noční spánek měřený polysomnografií nebyl použitím Provigilu ovlivněn.
Shift Work Sleep Disorder (SWSD)
Účinnost přípravku Provigil pro nadměrnou ospalost spojenou se SWSD byla prokázána v 12týdenní placebem kontrolované klinické studii. Celkem 209 pacientů s chronickou SWSD bylo randomizováno pro podávání Provigilu 200 mg / den nebo placeba. Všichni pacienti splnili kritéria Mezinárodní klasifikace poruch spánku (ICSD-10) pro chronickou SWSD (která jsou v souladu s kritérii DSM-IV Americké psychiatrické asociace pro poruchu spánku cirkadiánního rytmu: práce na směny Typ). Tato kritéria zahrnují 1) buď: a) primární stížnost na nadměrnou ospalost nebo nespavost, která je dočasně spojena s pracovní dobou (obvykle noční prací), která vyskytuje se během obvyklé spánkové fáze, nebo b) polysomnografie a MSLT prokazují ztrátu normálního vzoru spánek-bdění (tj. narušené chronobiologie) rytmičnost); a 2) žádné další lékařské nebo duševní poruchy nezohledňují příznaky a 3) symptomy nesplňují kritéria pro jakoukoli jinou poruchu spánku způsobující nespavost nebo nadměrnou ospalost (např. změna časového pásma [jet lag] syndrom).
Je třeba poznamenat, že ne všichni pacienti se stížností na ospalost, kteří se také zabývají prací na směny, splňují kritéria pro diagnózu SWSD. Do klinického hodnocení byli zařazeni pouze pacienti, kteří byli symptomatičtí po dobu nejméně 3 měsíců.
Registrovaní pacienti byli také povinni pracovat minimálně 5 nočních směn za měsíc, mít nadměrnou spavost čas jejich nočních směn (skóre MSLT <6 minut) a denní nespavost doložená denním polysomnogramem (PSG).
Primárními měřítky účinnosti byly 1) spánková latence, jak bylo stanoveno pomocí testu vícenásobné spánkové latence (MSLE) provedeného během simulované noční směny ve 12. týdnu nebo závěrečná návštěva a 2) změna celkového stavu onemocnění pacienta, měřeno pomocí klinického globálního zobrazení změny (CGI-C) ve 12. týdnu nebo poslední návštěva. U pacientů léčených přípravkem Provigil bylo ve srovnání s pacienty léčenými placebem prokázáno statisticky významné prodloužení doby do spánku ve srovnání s pacienty léčenými placebem [tabulka 1] (p <0,05). Zlepšení na CGI-C bylo také pozorováno jako statisticky významné (p <0,001). (Vidět Klinické stezky, Popis narkolepsie výše pro popis těchto testů.)
Denní spánek měřený polysomnografií nebyl použitím Provigilu ovlivněn.
HTML schránka
| Porucha | Opatření | Provigil 200 mg * |
Provigil 400 mg * |
Placebo | |||
| * Výrazně odlišné od placeba ve všech studiích (p <0,01 pro všechny studie, ale SWSD, což bylo p <0,05) | |||||||
| Základní hodnota | Změna od Baseline |
Základní hodnota | Změna od Baseline |
Základní hodnota | Změnit z Základní hodnota |
||
| Narkolepsie I. | MWT | 5.8 | 2.3 | 6.6 | 2.3 | 5.8 | -0.7 |
| Narkolepsie II | MWT | 6.1 | 2.2 | 5.9 | 2.0 | 6.0 | -0.7 |
| OSAHS | MWT | 13.1 | 1.6 | 13.6 | 1.5 | 13.8 | -1.1 |
| SWSD | MSLT | 2.1 | 1.7 | - | - | 2.0 | 0.3 |
| Porucha | Provigil 200 mg * |
Provigil 400 mg * |
Placebo |
| * Významně odlišné od placeba ve všech studiích (p <0,01) | |||
| Narkolepsie I. | 64% | 72% | 37% |
| Narkolepsie II | 58% | 60% | 38% |
| OSAHS | 61% | 68% | 37% |
| SWSD | 74% | - | 36% |
horní
Indikace a použití
Přípravek Provigil je indikován ke zlepšení bdělosti u dospělých pacientů s nadměrnou ospalostí spojenou s narkolepsií, obstrukčním spánkovým apnoe / hypopnoe syndromem a poruchou pracovního spánku.
V OSAHS je Provigil indikován jako doplněk ke standardní léčbě pro základní překážku. Pokud je léčba volby pro pacienta trvalým pozitivním tlakem dýchacích cest (CPAP), mělo by být před zahájením léčby přípravkem Provigil vynaloženo maximální úsilí k léčbě přípravkem CPAP po přiměřenou dobu. Pokud se Provigil používá doplňkově s CPAP, je nutné povzbuzovat a pravidelně vyhodnocovat dodržování CPAP.
Ve všech případech je nanejvýš důležitá pečlivá pozornost k diagnostice a léčbě základní poruchy spánku. Předepisující lékaři by si měli být vědomi toho, že někteří pacienti mohou mít více než jednu poruchu spánku, což přispívá k jejich nadměrné ospalosti.
Účinnost modafinilu při dlouhodobém užívání (více než 9 týdnů v klinických studiích s narkolepsií a 12 týdnů v klinických studiích OSAHS a SWSD) nebyla systematicky hodnocena u placebem kontrolovaných zkoušky. Lékař, který se rozhodne předepsat přípravek Provigil na delší dobu u pacientů s narkolepsií, OSAHS nebo SWSD, by měl pravidelně přehodnocovat dlouhodobou užitečnost pro jednotlivého pacienta.
horní
Kontraindikace
Provigil je kontraindikován u pacientů se známou přecitlivělostí na modafinil, armodafinil nebo jeho neaktivní složky.
horní
Varování
Vážná vyrážka, včetně Stevens-Johnsonova syndromu
U dospělých a dětí byla hlášena závažná vyrážka vyžadující hospitalizaci a přerušení léčby v souvislosti s používáním modafinilu.
Modafinil není schválen pro použití u pediatrických pacientů pro jakoukoli indikaci.
V klinických studiích s modafinilem byla incidence vyrážky, která vedla k přerušení léčby, přibližně 0,8% (13 na 1 585) u dětských pacientů (věk <17 let); tyto vyrážky zahrnovaly 1 případ možného Stevens-Johnsonova syndromu (SJS) a 1 případ zjevné hypersenzitivní reakce na více orgánů. Několik případů bylo spojeno s horečkou a dalšími abnormalitami (např. Zvracení, leukopenie). Střední doba do vyrážky, která vedla k přerušení léčby, byla 13 dní. U 380 pediatrických pacientů, kteří dostávali placebo, nebyly pozorovány žádné takové případy. V klinických studiích s dospělými (0 na 4 264) modafinilu nebyly hlášeny žádné závažné kožní vyrážky.
Vzácné případy závažných nebo život ohrožujících vyrážek, včetně SJS, toxické epidermální nekrolýzy (TEN) a vyrážky s drogami s Eosinofilie a systémové příznaky (DRESS) byly hlášeny u dospělých a dětí v celosvětovém postmarketingovém sledování Zkušenosti. Míra hlášení TEN a SJS spojená s používáním modafinilu, která je obecně považována za podceňovaná kvůli nedostatečnému hlášení, překračuje míru výskytu na pozadí. Odhady míry výskytu pozadí těchto závažných kožních reakcí v obecné populaci se pohybují mezi 1 až 2 případy na milion osob na rok.
Není známo, že by předpovídalo riziko výskytu nebo závažnost vyrážky spojené s modafinilem. Téměř všechny případy závažné vyrážky spojené s modafinilem se objevily během 1 až 5 týdnů po zahájení léčby. Byly však hlášeny izolované případy po dlouhodobé léčbě (např. 3 měsíce). V souladu s tím nelze na dobu trvání léčby spoléhat jako na předpovědi potenciálního rizika ohlašovaného prvním výskytem vyrážky.
Ačkoli se u modafinilu vyskytují také benigní vyrážky, není možné spolehlivě předpovědět, které vyrážky se ukáží jako závažné. Modafinil by proto měl být obvykle přerušen při prvních známkách vyrážky, pokud vyrážka zjevně nesouvisí s drogami. Přerušení léčby nemusí zabránit tomu, aby se vyrážka stala život ohrožující nebo natrvalo znemožňovala nebo znetvořovala.
Angioedémové a anafylaktoidní reakce
Jeden závažný případ angioedému a jeden případ přecitlivělosti (s vyrážkou, dysfagií a bronchospasmem) byly pozorováno u 1 595 pacientů léčených armodafinilem, R enantiomerem modafinilu (což je racemický) směs). V klinických studiích s modafinilem nebyly tyto případy pozorovány. Při postmarketingovém sledování s modafinilem byl však hlášen angioedém. Pacienti by měli být upozorněni, aby léčbu přerušili a okamžitě informovali svého lékaře o všech příznacích nebo symptomy naznačující angioedém nebo anafylaxi (např. otok obličeje, očí, rtů, jazyka nebo hrtan; potíže s polykáním nebo dýcháním; chrapot).
Reakce přecitlivělosti na více orgánů
Reakce přecitlivělosti na více orgánů, včetně alespoň jedné úmrtí při postmarketingovém sledování, mají došlo k těsnému časovému spojení (střední doba do detekce 13 dnů: rozmezí 4–33) do zahájení modafinil.
Přestože bylo hlášeno jen omezené množství zpráv, reakce přecitlivělosti na více orgánů mohou vést k hospitalizaci nebo mohou ohrozit život. Není známo, že by předpovídalo riziko výskytu nebo závažnost hypersenzitivních reakcí na více orgánů spojených s modafinilem. Příznaky a příznaky této poruchy byly různé; pacienti však obvykle, i když ne výhradně, vykazovali horečku a vyrážku spojenou s postižením jiných orgánů. Mezi další související projevy patřily myokarditida, hepatitida, abnormality testu jaterních funkcí, hematologické abnormality (např. eosinofilie, leukopenie, trombocytopenie), pruritus a astenie. Protože hypersenzitivita na více orgánů je různá ve své expresi, mohou nastat další příznaky a příznaky orgánového systému, které zde nejsou uvedeny.
Pokud je podezření na přecitlivělost na více orgánů, měla by být léčba přípravkem Provigil přerušena. Přestože neexistují žádné případy, které by naznačovaly zkříženou citlivost na jiné léky, které tento syndrom vyvolávají, zkušenosti s drogami spojenými s přecitlivělostí na více orgánů by naznačovaly, že se jedná o a možnost.
Trvalá ospalost
Pacienti s neobvyklou mírou ospalosti, kteří užívají přípravek Provigil, by měli být upozorněni, že se jejich úroveň bdělosti nemusí vrátit k normálu. Pacienti s nadměrnou spavostí, včetně těch, kteří užívají Provigil, by měli být často přehodnocováni stupeň ospalosti a případně se doporučuje vyhnout se řízení nebo jakékoli jiné potenciálně nebezpečné činnosti. Předepisující lékaři by si měli být rovněž vědomi toho, že pacienti nemusí uznat ospalost nebo ospalost, dokud se během konkrétních činností přímo nezeptají na ospalost nebo ospalost.
Psychiatrické příznaky
U pacientů léčených modafinilem byly hlášeny psychiatrické nežádoucí účinky. Po uvedení přípravku na trh byly nežádoucí účinky spojené s užíváním modafinilu zahrnuty mánie, bludy, halucinace, sebevražedné myšlenky a agrese, z nichž některé vedly k hospitalizaci. Mnoho, ale ne všichni, měli předchozí psychiatrickou anamnézu. Jeden zdravý dobrovolník mužského pohlaví vyvinul myšlenky na reference, paranoidní bludy a sluchové halucinace ve spojení s opakovanými 600 mg dávkami modafinilu a deprivací spánku. 36 hodin po přerušení léčby nebyl prokázán psychóza.
V databázi kontrolovaných studií modafinilu u dospělých byly psychiatrické příznaky, které vedly k přerušení léčby (s frekvencí> 0,3%) a častěji hlášeny u pacientů léčených modafinilem ve srovnání s placebem byla úzkost (1%), nervozita (1%), nespavost (<1%), zmatenost (<1%), agitace (<1%) a deprese (<1%). Pokud je přípravek Provigil podáván pacientům s anamnézou psychózy, deprese nebo mánie v anamnéze, je třeba postupovat opatrně. U pacientů léčených přípravkem Provigil je třeba zvážit možný výskyt nebo zhoršení psychiatrických příznaků. Pokud se psychiatrické příznaky objeví ve spojení s podáváním přípravku Provigil, zvažte ukončení léčby přípravkem Provigil.
horní
Opatření
Diagnostika poruch spánku
Přípravek Provigil by měl být používán pouze u pacientů, u nichž bylo provedeno úplné vyhodnocení jejich nadměrné ospalosti a u nichž diagnóza narkolepsie, OSAHS a / nebo SWSD byla stanovena v souladu s diagnostickými kritérii ICSD nebo DSM (viz Klinické stezky). Takové hodnocení obvykle sestává z úplné historie a fyzického vyšetření a může být doplněno testováním v laboratorním prostředí. Někteří pacienti mohou mít více než jednu poruchu spánku přispívající k jejich nadměrné ospalosti (např. OSAHS a SWSD shodné u stejného pacienta).
Všeobecné
Přestože se ukázalo, že modafinil nevyvolává funkční poškození, může jakýkoli lék ovlivňující CNS změnit úsudek, myšlení nebo motorické schopnosti. Pacienti by měli být obeznámeni s obsluhou automobilu nebo jiného nebezpečného strojního zařízení, dokud nejsou přiměřeně jisté, že terapie Provigilem nepříznivě neovlivní jejich schopnost zapojit se do takových činnosti.
Použití CPAP u pacientů s OSAHS
V OSAHS je Provigil indikován jako doplněk ke standardní léčbě pro základní překážku. Pokud je léčba volby pro pacienta trvalým pozitivním tlakem dýchacích cest (CPAP), mělo by být před zahájením léčby přípravkem Provigil vynaloženo maximální úsilí k léčbě přípravkem CPAP po přiměřenou dobu. Pokud se Provigil používá doplňkově s CPAP, je nutné povzbuzovat a pravidelně vyhodnocovat dodržování CPAP.
Kardiovaskulární systém
Modafinil nebyl hodnocen u pacientů s nedávným anamnézou infarktu myokardu nebo nestabilní anginy pectoris, a takoví pacienti by měli být léčeni opatrně.
V klinických studiích s přípravkem Provigil se projevy a příznaky včetně bolesti na hrudi, bušení srdce, dušnosti a přechodné ischemické choroby Změny T-vln na EKG byly pozorovány u tří subjektů ve spojení s prolapsem mitrální chlopně nebo levé komory hypertrofie. Doporučuje se, aby tablety Provigilu nebyly používány u pacientů s anamnézou hypertrofie levé komory nebo v anamnéze pacienti s prolapsem mitrální chlopně, kteří zažili syndrom prolapsu mitrální chlopně při předchozím podávání CNS stimulanty. Takové příznaky mohou zahrnovat, ale nejsou omezeny na ischemické změny EKG, bolest na hrudi nebo arytmii. Pokud se objeví jakýkoli z těchto příznaků, zvažte srdeční vyšetření.
Monitorování krevního tlaku v krátkodobých (<3 měsíců) kontrolovaných studiích neprokázalo žádné klinicky významné změny průměrného systolického a diastolického krevního tlaku u pacientů užívajících Provigil ve srovnání s placebo. Retrospektivní analýza použití antihypertenziv v těchto studiích však ukázala, že větší podíl pacienti na Provigilu vyžadovali nové nebo zvýšené používání antihypertenziv (2,4%) ve srovnání s pacienty na placebu (0,7%). Rozdílné použití bylo o něco větší, když byly zahrnuty pouze studie OSAHS, se 3,4% pacientů Provigil a 1,1% pacientů na placebu vyžadujících takové změny v používání antihypertenziv léky. U pacientů na přípravku Provigil může být vhodné zvýšené monitorování krevního tlaku.
Pacienti užívající steroidní antikoncepci
Účinnost steroidních kontraceptiv může být snížena při použití s tabletami Provigil a po dobu jednoho měsíce po ukončení léčby (viz Opatření, Lékové interakce). U pacientů léčených tabletami Provigilu a jeden měsíc po ukončení léčby přípravkem Provigil se doporučuje alternativní nebo souběžné metody antikoncepce.
Pacienti užívající cyklosporin
Hladiny cyklosporinu v krvi mohou být při použití s přípravkem Provigil sníženy (viz Opatření, Lékové interakce). Při současném užívání těchto léků by mělo být zváženo monitorování koncentrací cirkulujícího cyklosporinu a odpovídající úprava dávkování cyklosporinu.
Pacienti se závažným poškozením jater
U pacientů se závažným poškozením jater, s cirhózou nebo bez cirhózy (viz Klinická farmakologie), Provigil by měl být podáván ve snížené dávce (viz Dávkování a správa).
Pacienti se závažným poškozením ledvin
Pro určení bezpečnosti a účinnosti dávkování u pacientů se závažným poškozením ledvin nejsou k dispozici dostatečné informace. (Farmakokinetika při poškození ledvin viz Klinická farmakologie.)
Starší pacienti
U starších pacientů může být v důsledku stárnutí eliminace modafinilu a jeho metabolitů snížena. Proto by se mělo u této populace zvážit použití nižších dávek. (Vidět Klinická farmakologie a Dávkování a správa).
Informace pro pacienty
Lékařům se doporučuje diskutovat o následujících problémech s pacienty, kterým předepisují přípravek Provigil.
Přípravek Provigil je indikován u pacientů, kteří mají abnormální úroveň ospalosti. Bylo prokázáno, že se Provigil zlepšuje, ale neodstraní tuto neobvyklou tendenci usnout. Pacienti by proto neměli měnit své předchozí chování, pokud jde o potenciálně nebezpečné činnosti (např. Řízení vozidla, obsluha strojů) nebo jiné činnosti vyžadující přiměřenou úroveň bdělosti, dokud a pokud není prokázáno, že by léčba Provigilem vyvolala takové úrovně bdělosti, které takové činnosti. Pacienti by měli být upozorněni, že přípravek Provigil není náhradou spánku.
Pacienti by měli být informováni o tom, že může být důležité, aby pokračovali ve své dříve předepsané léčbě (např. Pacienti s OSAHS, kteří dostávají CPAP, by tak měli i nadále).
Pacienti by měli být informováni o dostupnosti příbalové informace pro pacienty a měli by být instruováni, aby si před použitím přípravku Provigil přečetli příbalovou informaci.
Pacientům by mělo být doporučeno, aby se obrátili na svého lékaře, pokud se u nich objeví bolest na hrudi, vyrážka, deprese, úzkost nebo známky psychózy nebo mánie.
Těhotenství
Pacientky by měly být upozorněny, aby informovaly svého lékaře, pokud během léčby otěhotní nebo mají v úmyslu otěhotnět. Při používání steroidních antikoncepčních prostředků (včetně depotních nebo implantovatelných) by měla být pacienti opatrně upozorněni na možné zvýšené riziko těhotenství antikoncepční prostředky) s přípravkem Provigil a jeden měsíc po ukončení léčby (viz Karcinogeneze, mutageneze, zhoršení plodnosti a Těhotenství).
Ošetřovatelství
Pacientům by mělo být doporučeno, aby informovali svého lékaře, pokud kojí dítě.
Souběžné léky
Pacientům by mělo být doporučeno, aby informovali svého lékaře, pokud užívají nebo plánují nějaké užívat léky na předpis nebo volně prodejné léky, protože existuje potenciál pro interakce mezi Provigilem a jiné drogy.
Alkohol
Pacienti by měli být upozorněni, že použití přípravku Provigil v kombinaci s alkoholem nebylo studováno. Pacienti by měli být upozorněni, že je rozumné vyhýbat se alkoholu během užívání přípravku Provigil.
Alergické reakce
Pacientům by mělo být doporučeno, aby přestali užívat Provigil a informovali svého lékaře, pokud se u nich objeví vyrážka, kopřivka, bolest v ústech, puchýře, loupání kůže, potíže s polykáním nebo dýcháním nebo související alergie jev.
Lékové interakce
CNS aktivní léky
Methylfenidát
Ve studii s jednou dávkou u zdravých dobrovolníků bylo současné podávání modafinilu (200 mg) s methylfenidát (40 mg) nezpůsobil žádné významné změny ve farmakokinetice ani jedné lék. Absorpce Provigilu však může být při současném podávání s methylfenidátem zpožděna přibližně o jednu hodinu.
Ve studii se zdravými dobrovolníky ve více dávkách v ustáleném stavu byl modafinil podáván jednou denně v dávce 200 mg / den po dobu 7 dnů a poté 400 mg / den po dobu 21 dnů. Podávání methylfenidátu (20 mg / den) během 22-28 dnů léčby modafinilem 8 hodin po podání denní dávka modafinilu nezpůsobila žádné významné změny ve farmakokinetice modafinil.
Dextroamfetamin
Ve studii s jednou dávkou u zdravých dobrovolníků bylo současné podávání modafinilu (200 mg) s dextroamfetamin (10 mg) nezpůsobil žádné významné změny ve farmakokinetice ani jednoho lék. Absorpce Provigilu však může být při současném podávání s dextroamfetaminem zpožděna přibližně o jednu hodinu.
Ve studii se zdravými dobrovolníky ve více dávkách v ustáleném stavu byl modafinil podáván jednou denně v dávce 200 mg / den po dobu 7 dnů a poté 400 mg / den po dobu 21 dnů. Podávání dextroamfetaminu (20 mg / den) během 22-28 dnů léčby modafinilem 7 hodin po podání denní dávka modafinilu nezpůsobila žádné významné změny ve farmakokinetice modafinil.
Klomipramin
Souběžné podání jedné dávky klomipraminu (50 mg) během prvních tří dnů léčby modafinil (200 mg / den) u zdravých dobrovolníků neprokázal účinek na farmakokinetiku ani jednoho léčiva. U pacientů s narkolepsií během léčby modafinilem byl však zaznamenán jeden případ zvýšených hladin klomipraminu a jeho aktivního metabolitu desmethylclomipraminu.
Triazolam
Ve studii lékové interakce mezi Provigilem a ethinylestradiolem (EE2), ve stejné dny jako pro při plazmatickém odběru pro farmakokinetiku EE2 byla také podána jediná dávka triazolamu (0,125 mg). Střední Cmax a AUC0-β triazolamu byly sníženy o 42%, respektive 59%, a jeho eliminační poločas byl snížen přibližně o hodinu po léčbě modafinilem.
Inhibitory monoaminooxidázy (MAO)
Interakční studie s inhibitory monoaminooxidázy nebyly provedeny. Při současném podávání inhibitorů MAO a modafinilu je proto třeba postupovat opatrně.
Další léky
Warfarin
Ve farmakokinetických profilech R- a S-warfarinu u zdravých jedinců při jednorázové dávce racemického warfarinu (5 mg) nedošlo k žádným významným změnám. po chronickém podání modafinilu (200 mg / den po dobu 7 dnů, poté 400 mg / den po dobu 27 dnů) vzhledem k profilům u subjektů placebo. Doporučuje se však častější sledování protrombinových časů / INR, kdykoli je přípravek Provigil podáván společně s warfarinem (viz Klinická farmakologie, Farmakokinetika, lékové interakce).
Ethinylestradiol
Podávání modafinilu dobrovolnicím žen jednou denně v dávce 200 mg / den po dobu 7 dnů, po nichž následovalo 400 mg / den po dobu 21 dnů vedlo k průměrnému 11% snížení Cmax a 18% snížení AUC0-24 ethinylestradiolu (EE2; 0,035 mg; podáván orálně s norgestimátem). Nebyla patrná žádná změna v rychlosti eliminace ethinylestradiolu.
Cyklosporin
Jeden případ interakce mezi modafinilem a cyklosporinem, substrátem CYP3A4, byl hlášen u 41leté ženy, která podstoupila transplantaci orgánů. Po jednom měsíci podání 200 mg / den modafinilu byly hladiny cyklosporinu v krvi sníženy o 50%. Předpokládalo se, že interakce je způsobena zvýšeným metabolismem cyklosporinu, protože se nezměnil žádný další faktor, který by mohl ovlivnit dispozici léčiva. Může být nutné upravit dávkování cyklosporinu.
Potenciální interakce s léky, které inhibují, indukují nebo jsou metabolizovány izoenzymy cytochromu P-450 a jinými hepatickými enzymy
Ve studiích in vitro s použitím primárních kultur lidských hepatocytů bylo prokázáno, že modafinil mírně indukuje CYP1A2, CYP2B6 a CYP3A4 v závislosti na koncentraci. Ačkoli výsledky indukce založené na experimentech in vitro nemusí nutně predikovat odpověď in vivo, opatrnost je třeba uplatňovat, když se Provigil podává společně s léky, které pro ně závisí na těchto třech enzymech odbavení. Konkrétně by mohlo dojít k nižším hladinám těchto léků v krvi (viz Další léky, Cyclosporinea výše).
Expozice lidských hepatocytů modafinilu in vitro vyvolala zjevné koncentrační potlačení exprese aktivity CYP2C9 což naznačuje, že existuje potenciál pro metabolickou interakci mezi modafinilem a substráty tohoto enzymu (např. S-warfarin a fenytoin). V následné klinické studii u zdravých dobrovolníků chronická léčba modafinilem neprokázala významný účinek na farmakokinetiku jedné dávky warfarinu ve srovnání s placebem (viz Opatření, Drug Interactions, Warfarin).
Studie in vitro s použitím lidských jaterních mikrozomů ukázaly, že modafinil reverzibilně inhiboval CYP2C19 ve farmakologicky relevantních koncentracích modafinilu. CYP2C19 je také reverzibilně inhibován, s podobnou účinností, cirkulujícím metabolitem, modafinil sulfonem. Přestože maximální koncentrace modafinil sulfonu v plazmě jsou mnohem nižší než u mateřských modafinil, kombinovaný účinek obou sloučenin by mohl vést k trvalé částečné inhibici enzym. Léky, které jsou do značné míry vylučovány metabolismem CYP2C19, jako je diazepam, propranolol, fenytoin (také prostřednictvím CYP2C9) nebo S-mefenytoin může být eliminován při současném podání s přípravkem Provigil a může vyžadovat snížení dávky a monitorování pro toxicitu.
Tricyklická antidepresiva
CYP2C19 také poskytuje pomocnou cestu pro metabolismus určitých tricyklických antidepresiv (např. Klomipraminu a desipraminu), které jsou primárně metabolizovány CYP2D6. U tricyklicky léčených pacientů s deficitem CYP2D6 (tj. U těch, kteří jsou špatnými metabolizátory debrisoquinu; 7-10% bělošské populace; podobné nebo nižší v jiných populacích), může být množství metabolismu CYP2C19 podstatně zvýšeno. Provigil může v této podskupině pacientů způsobit zvýšení hladiny tricyklických látek. Lékaři by si měli být vědomi toho, že u těchto pacientů může být nutné snížit dávku tricyklických látek.
Kromě toho, kvůli částečnému zapojení CYP3A4 do metabolické eliminace modafinilu, společné podávání silných induktorů CYP3A4 (např. karbamazepin, fenobarbital, rifampin) nebo inhibitory CYP3A4 (např. ketokonazol, itrakonazol) by mohly změnit plazmatické hladiny modafinil.
Karcinogeneze, mutageneze, zhoršení plodnosti
Karcinogeneze
Byly provedeny studie karcinogenity, ve kterých byl modafinil podáván ve stravě myším po dobu 78 týdnů a potkanům po dobu 104 týdnů v dávkách 6, 30 a 60 mg / kg / den. Nejvyšší studovaná dávka je 1,5 (myš) nebo 3 (krysa) krát větší než doporučená denní dávka člověka modafinilu (200 mg) pro dospělého člověka na bázi mg / m2. V těchto studiích nebyl prokázán tumorigeneze spojený s podáváním modafinilu. Protože však studie na myších používala nedostatečnou vysokou dávku, která nebyla reprezentativní pro maximální tolerovanou dávku, byla v Tg provedena následná studie karcinogenity. AC transgenní myš. Dávky vyhodnocené v Tg. Test AC byl 125, 250 a 500 mg / kg / den, dermálně. Nebyl prokázán tumorigenicita spojená s podáváním modafinilu; tento dermální model však nemusí dostatečně posoudit karcinogenní potenciál perorálně podaného léčiva.
Mutageneze
Modafinil neprokázal žádný důkaz mutagenního nebo klastogenního potenciálu v řadě in vitro (tj. Test bakteriální reverzní mutace, test myšího lymfomu tk, chromozomální aberace) test na lidských lymfocytech, test na transformaci buněk v myších embryích BALB / 3T3) testy v nepřítomnosti nebo v přítomnosti metabolické aktivace nebo in vivo (mikronukleus kostní dřeně myší) testy. Modafinil byl také negativní v testu neplánované syntézy DNA v hepatocytech potkanů.
Snížení plodnosti
Perorální podání modafinilu (dávky až 480 mg / kg / den) samcům a samicím potkanů před a v průběhu páření a pokračování u žen až do 7. dne těhotenství vedlo ke zvýšení času na párování na nejvyšší úrovni dávka; nebyly pozorovány žádné účinky na jiné parametry plodnosti nebo reprodukce. Dávka bez účinku 240 mg / kg / den byla spojena s plazmatickou expozicí modafinilu (AUC) přibližně stejnou jako u lidí při doporučené dávce 200 mg.
Těhotenství
Kategorie těhotenství C:
Ve studiích prováděných na potkanech a králících byla pozorována vývojová toxicita při klinicky relevantních expozicích.
Modafinil (50, 100 nebo 200 mg / kg / den) podávaný orálně těhotným potkanům po celou dobu vyvolané organogeneze, v nepřítomnosti mateřská toxicita, zvýšení resorpcí a zvýšený výskyt viscerálních a kosterních variací u potomků na nejvyšší úrovni dávka. Vyšší dávka bez účinku pro embryofetální vývojovou toxicitu u potkanů byla spojena s plazmou expozice modafinilu přibližně 0,5násobek AUC u lidí při doporučené denní dávce (RHD) 200 mg. V následné studii až do 480 mg / kg / den (expozice modafinilu v plazmě přibližně 2násobek AUC u lidí na RHD) však nebyly pozorovány žádné nepříznivé účinky na embryofetální vývoj.
Modafinil podávaný orálně těhotným králíkům v období organogeneze v dávkách 45, 90 a 180 mg / kg / den zvýšilo výskyt strukturálních změn plodu a úmrtí embryí při nejvyšší dávce. Nejvyšší dávka bez účinku pro vývojovou toxicitu byla spojena s plazmatickou AUC modafinilu přibližně stejnou jako AUC u lidí na RHD.
Perorální podání armodafinilu (R-enantiomer modafinilu; 60, 200 nebo 600 mg / kg / den) u březích potkanů po celou dobu organogeneze vedlo ke zvýšenému výskytu fetální viscerální a kosterní variace při střední dávce nebo vyšší a snížená tělesná hmotnost plodu na nejvyšší dávka. Dávka bez účinku pro embryofetální vývojovou toxicitu u potkanů byla spojena s plazmatickým Armodafinilem expozice (AUC) přibližně desetinásobek AUC pro armodafinil u lidí léčených modafinilem na RHD.
Podávání modafinilu potkanům během březosti a laktace při perorálních dávkách až 200 mg / kg / den vedlo ke snížení životaschopnost potomstva při dávkách vyšších než 20 mg / kg / den (AUC plazmatického modafinilu přibližně 0,1násobek AUC u lidí na RHD). U přežívajících potomků nebyly pozorovány žádné účinky na postnatální vývojové a neurobehaviorální parametry.
Neexistují dostatečné a dobře kontrolované studie u těhotných žen. Ve spojení s armodafinilem a modafinilem byly hlášeny dva případy retardace intrauterinního růstu a jeden případ spontánního potratu. Ačkoli farmakologie modafinilu a armodafinilu není identická s farmakologií sympatomimetických aminů, sdílí s touto třídou některé farmakologické vlastnosti. Některé z těchto léků byly spojeny s intrauterinní retardací růstu a spontánními potraty. Není známo, zda hlášené případy souvisejí s drogami.
Modafinil by měl být používán během těhotenství, pouze pokud potenciální přínos odůvodňuje možné riziko pro plod.
Práce a dodávky
Účinek modafinilu na porodu a porod u lidí nebyl systematicky zkoumán.
Kojící matky
Není známo, zda se modafinil nebo jeho metabolity vylučují do mateřského mléka. Protože se mnoho léků vylučuje do mateřského mléka, je třeba při podávání tablet přípravku Provigil kojící ženě postupovat opatrně.
Pediatrické použití
Bezpečnost a účinnost u pediatrických pacientů mladších 16 let nebyla stanovena. S použitím modafinilu u dětských pacientů byly spojeny závažné kožní vyrážky, včetně erythema multiforme major (EMM) a Stevens-Johnsonova syndromu (SJS). Varování, Serious Rash, včetně Stevens-Johnsonova syndromu).
V kontrolované 6týdenní studii bylo 165 pediatrických pacientů (ve věku 5-17 let) s narkolepsií léčeno modafinilem (n = 123) nebo placebem (n = 42). Nebyly zjištěny žádné statisticky významné rozdíly, které by upřednostňovaly modafinil před placebem při prodloužení latence spánku jako měřeno pomocí MSLT nebo ve vnímání ospalosti, jak bylo stanoveno na stupnici klinického globálního dojmu-klinika (CGI-C).
V kontrolovaných a otevřených klinických studiích se objevily nežádoucí účinky psychiatrického a nervového systému zahrnoval Touretteův syndrom, nespavost, nepřátelství, zvýšenou kataplexii, zvýšené hypnagogické halucinace a sebevraždu myšlenka. Byla také pozorována přechodná leukopenie, která ustoupila bez lékařského zásahu. V kontrolované klinické studii se u 3 z 38 dívek ve věku 12 let a starších léčených modafinilem vyskytla dysmenorea v porovnání s 0 z 10 dívek, které dostávaly placebo.
Geriatrické použití
Bezpečnost a účinnost u osob starších 65 let nebyla stanovena. Zkušenosti s omezeným počtem pacientů, kteří byli v klinických studiích starší 65 let, prokázali výskyt nežádoucích účinků podobných jiným věkovým skupinám.
horní
Nežádoucí reakce
Bezpečnost přípravku Modafinil byla hodnocena u více než 3500 pacientů, z nichž více než 2000 pacientů s nadměrným množstvím ospalost spojená s primárními poruchami spánku a bdělosti byla podána alespoň jedna dávka modafinil. V klinických studiích bylo zjištěno, že modafinil je obecně dobře tolerován a většina nežádoucích účinků byla mírná až střední.
Nejčastěji pozorované nežádoucí účinky (± 5%) spojené s používáním Provigilu častěji než u pacientů léčených placebem u placebem kontrolovaných klinickými studiemi primárních poruch spánku a bdělosti byly bolesti hlavy, nevolnost, nervozita, rýma, průjem, bolesti zad, úzkost, nespavost, závratě a dyspepsie. Profil nežádoucích účinků byl v těchto studiích podobný.
V placebem kontrolovaných klinických studiích 74 z 934 pacientů (8%), kteří dostávali Provigil, ukončilo léčbu kvůli nežádoucím účinkům ve srovnání s 3% pacientů, kteří dostávali placebo. Nejčastější důvody přerušení, které se vyskytly u Provigilu vyšší rychlostí než u placeba pacienti byli bolest hlavy (2%), nevolnost, úzkost, závratě, nespavost, bolest na hrudi a nervozita (každý <1%). V kanadské klinické studii byl 35letý obézní narkoleptik s předchozí anamnézou synkopálních epizod po 9 dnech léčby modafinilem došlo k 9-sekundové epizodě asystoly (rozděleno 300 mg / den) dávky).
Incidence v kontrolovaných pokusech
Následující tabulka (tabulka 3) představuje nežádoucí účinky, které se vyskytly v míře 1% nebo více a byly častější u dospělých pacientů léčených Provigilem než u pacientů léčených placebem v hlavní klinicky kontrolované placebem zkoušky.
Předpisující lékař by si měl být vědom toho, že níže uvedené údaje nelze použít k predikci frekvence nežádoucích účinků v EU průběhu obvyklé lékařské praxe, kde se charakteristiky pacienta a další faktory mohou lišit od těch, které se vyskytují během klinického vývoje studie. Podobně citované frekvence nemohou být přímo srovnávány s čísly získanými z jiných klinických zkoušek zahrnujících různé ošetření, použití nebo vyšetřovatele. Přezkum těchto frekvencí však předepisujícím poskytuje základ pro odhad relativního příspěvku lékových a nedrogových faktorů k výskytu nežádoucích účinků ve studované populaci.
| Systém těla | Preferovaný termín | Modafinil (n = 934) |
Placebo (n = 567) |
| * Šest dvojitě slepých, placebem kontrolovaných klinických studií s narkolepsií, OSAHS a SWSD. 1 Zahrnuty jsou události hlášené nejméně 1% pacientů léčených Provigilem, které byly častější než ve skupině s placebem; incidence je zaokrouhlena na nejbližší 1%. Terminologie nepříznivých zkušeností je kódována pomocí standardně upraveného slovníku COSTART. Události, u nichž byl výskyt Provigilu nejméně 1%, ale stejný nebo menší než placebo, nejsou v tabulce uvedeny. Tyto události zahrnovaly následující: infekce, bolest, náhodné zranění, bolest břicha, podchlazení, alergická reakce, astenie, horečka, virová infekce, bolest krku, migréna, abnormální elektrokardiogram, hypotenze, porucha zubů, zvracení, periodontální absces, zvýšená chuť k jídlu, ekchymóza, hyperglykémie, periferní edém, úbytek hmotnosti, hmotnost zisk, myalgie, křeče dolních končetin, artritida, kataplexie, poruchy myšlení, porucha spánku, zvýšený kašel, sinusitida, dušnost, bronchitida, vyrážka, konjunktivitida, bolest ucha, dysmenorea4, Infekce močových cest. 2 Zvýšené jaterní enzymy. 3 Orofaciální dyskineze. 4 Incidence upravená podle pohlaví. | |||
| Tělo jako celek | Bolest hlavy | 34% | 23% |
| Bolesti zad | 6% | 5% | |
| Chřipkový syndrom | 4% | 3% | |
| Bolest na hrudi | 3% | 1% | |
| Zimnice | 1% | 0% | |
| Tuhost krku | 1% | 0% | |
| Kardiovaskulární | Hypertenze | 3% | 1% |
| Tachykardie | 2% | 1% | |
| Palpitace | 2% | 1% | |
| Vasodilatace | 2% | 0% | |
| Zažívací | Nevolnost | 11% | 3% |
| Průjem | 6% | 5% | |
| Dyspepsie | 5% | 4% | |
| Suchá ústa | 4% | 2% | |
| Anorexie | 4% | 1% | |
| Zácpa | 2% | 1% | |
| Abnormální funkce jater2 | 2% | 1% | |
| Nadýmání | 1% | 0% | |
| Ulcerace v ústech | 1% | 0% | |
| Žízeň | 1% | 0% | |
| Hemic / Lymfatický | Eosinofilie | 1% | 0% |
| Metabolické / nutriční | Otok | 1% | 0% |
| Nervový | Nervozita | 7% | 3% |
| Nespavost | 5% | 1% | |
| Úzkost | 5% | 1% | |
| Závrať | 5% | 4% | |
| Deprese | 2% | 1% | |
| Parestézie | 2% | 0% | |
| Spavost | 2% | 1% | |
| Hypertonie | 1% | 0% | |
| Dyskineze3 | 1% | 0% | |
| Hyperkinezie | 1% | 0% | |
| Míchání | 1% | 0% | |
| Zmatek | 1% | 0% | |
| Třes | 1% | 0% | |
| Emoční schopnost | 1% | 0% | |
| Závrať | 1% | 0% | |
| Respirační | Rýma | 7% | 6% |
| Zánět hltanu | 4% | 2% | |
| Porucha plic | 2% | 1% | |
| Epistaxe | 1% | 0% | |
| Astma | 1% | 0% | |
| Kůže / dodatky | Pocení | 1% | 0% |
| Herpes Simplex | 1% | 0% | |
| Speciální smysly | Amblyopie | 1% | 0% |
| Abnormální vize | 1% | 0% | |
| Chuť perverze | 1% | 0% | |
| Bolest očí | 1% | 0% | |
| Urogenital | Abnormalita moči | 1% | 0% |
| Hematurie | 1% | 0% | |
| Pyuria | 1% | 0% |
Závislost na dávce nežádoucích účinků
V dospělých placebem kontrolovaných klinických studiích, které porovnávaly dávky 200, 300 a 400 mg / den Provigil a placebo, jediné nežádoucí účinky, které jasně souvisely s dávkou, byly bolesti hlavy a úzkost.
Změny vitálních funkcí
Zatímco nedošlo k žádné stálé změně průměrných hodnot srdeční frekvence nebo systolického a diastolického krevního tlaku, požadavek na antihypertenzivum byl mírně vyšší u pacientů na Provigilu ve srovnání s placebem (viz Opatření).
Změny hmotnosti
V placebem kontrolovaných klinických studiích nebyly klinicky významné rozdíly ve změně tělesné hmotnosti u pacientů léčených Provigilem ve srovnání s pacienty léčenými placebem.
Laboratorní změny
Ve studiích fáze 1, 2 a 3 byly sledovány parametry klinické chemie, hematologie a analýzy moči. V těchto studiích bylo zjištěno, že průměrné plazmatické hladiny gama glutamyltransferázy (GGT) a alkalické fosfatázy (AP) byly po podání Provigilu vyšší, ale nikoli placebo. Jen málo subjektů však mělo zvýšení GGT nebo AP mimo normální rozsah. Zdá se, že posuny k vyšším, ale nikoli klinicky významně abnormálním, hodnotám GGT a AP v čase v populaci léčené Provigilem vzrostly v klinických studiích fáze 3. Nebyly patrné žádné rozdíly v alaninaminotransferáze, aspartátaminotransferáze, celkovém proteinu, albuminu nebo celkovém bilirubinu.
Změny EKG
V placebem kontrolovaných klinických studiích po podání Provigilu nebyl nalezen žádný výskyt abnormalit EKG, který by nastal po léčbě.
Postmarketingové zprávy
Následující nežádoucí účinky byly zjištěny při použití přípravku Provigil po schválení. Protože tyto reakce jsou uváděny dobrovolně z populace nejisté velikosti, není možné spolehlivě odhadnout jejich frekvenci nebo stanovit příčinnou souvislost s expozicí léčivu. Rozhodnutí o zahrnutí těchto reakcí do označování jsou obvykle založena na jednom nebo více z následujících faktorů: 1) závažnost reakce, 2) četnost hlášení nebo 3) příčinná souvislost s Provigil.
Hematologická: agranulocytóza
horní
Zneužívání drog a závislost
Třída kontrolovaných látek
Modafinil (Provigil) je uveden v seznamu IV zákona o kontrolovaných látkách.
Potenciál zneužití a závislost
Kromě svého účinku na podporu bdělosti a zvýšené pohybové aktivity u zvířat, u lidí, Provigil produkuje psychoaktivní a euforické účinky, změny nálady, vnímání, myšlení a pocity typické pro jiné stimulanty CNS. V in vitro vazebných studiích se modafinil váže na místo zpětného vychytávání dopaminu a způsobuje zvýšení extracelulárního dopaminu, ale nezvyšuje uvolňování dopaminu. Modafinil posiluje, o čemž svědčí jeho vlastní podání u opic, které byly dříve vyškoleny k vlastnímu podávání kokainu. V některých studiích byl modafinil také částečně diskriminován jako stimulant. Lékaři by měli pečlivě sledovat pacienty, zejména ty, kteří mají v anamnéze zneužívání drog a / nebo stimulantů (např. Methylfenidát, amfetamin nebo kokain). Pacienti by měli být sledováni na známky zneužití nebo zneužití (např. Zvýšení dávek nebo chování při hledání drog).
Potenciál zneužití modafinilu (200, 400 a 800 mg) byl hodnocen ve srovnání s methylfenidátem (45 a 90 mg) v lůžkové studii u osob se zkušenostmi se zneužíváním drog. Výsledky této klinické studie prokázaly, že modafinil vyvolal psychoaktivní a euforické účinky a pocity shodné s jinými plánovanými stimulanty CNS (methylfenidát).
Vybrání
Účinky stažení modafinilu byly sledovány po 9 týdnech používání modafinilu v jedné klinické studii s kontrolovanou fází 3 v USA. Během 14 dnů pozorování nebyly pozorovány žádné specifické příznaky abstinenčního stavu, u narkoleptických pacientů se však vrátila ospalost.
horní
Předávkování
Lidské zkušenosti
V klinických studiích bylo celkem 151 dávek specifikovaných v protokolu v rozmezí od 1 000 do 1 600 mg / den (5 až 8násobek doporučené denní dávky 200 mg) bylo podáno 32 subjektům, včetně 13 subjektů, které dostávaly dávky 1 000 nebo 1 200 mg / den po dobu 7 až 21 po sobě jdoucích dny. Kromě toho došlo k několika záměrným akutním předávkování; dva největší z nich jsou 4500 mg a 4 000 mg užívané dvěma subjekty účastnícími se studií zahraniční deprese. Žádný z těchto studovaných subjektů nezaznamenal neočekávané nebo život ohrožující účinky. Nežádoucí účinky, které byly hlášeny při těchto dávkách, zahrnovaly excitaci nebo rozrušení, nespavost a mírné nebo střední zvýšení hemodynamických parametrů. Mezi další pozorované účinky vysokých dávek v klinických studiích patří úzkost, podrážděnost, agresivita, zmatenost, nervozita, třes, palpitace, poruchy spánku, nevolnost, průjem a snížený protrombin čas.
Z postmarketingových zkušeností nebyly hlášeny žádné fatální předávkování zahrnující modafinil samotný (dávky až 12 gramů). Předávkování zahrnující více léků, včetně modafinilu, mělo za následek fatální následky. Mezi příznaky, které nejčastěji doprovázejí předávkování modafinilem, samostatně nebo v kombinaci s jinými léky, patří: nespavost; příznaky centrálního nervového systému, jako je neklid, dezorientace, zmatenost, excitace a halucinace; zažívací změny, jako je nauzea a průjem; a kardiovaskulární změny, jako je tachykardie, bradykardie, hypertenze a bolest na hrudi.
U dětí ve věku 11 měsíců byly hlášeny případy náhodného požití / předávkování. Nejvyšší hlášené náhodné požití na bázi mg / kg se vyskytlo u tříletého chlapce, který požil 800-1000 mg (50-63 mg / kg) modafinilu. Dítě zůstalo stabilní. Příznaky spojené s předávkováním u dětí byly podobné příznakům pozorovaným u dospělých.
Řízení předávkování
Dosud nebylo identifikováno žádné specifické antidotum proti toxickým účinkům předávkování modafinilem. Takové předávkování by mělo být řešeno především podpůrnou péčí, včetně kardiovaskulárního sledování. Nejsou-li kontraindikace, je třeba zvážit vyvolané zvracení nebo výplach žaludku. Neexistují žádné údaje, které by naznačovaly užitečnost dialýzy nebo okyselení moči nebo alkalizace při zvýšení eliminace léčiva. Lékař by měl zvážit kontakt s centrem pro kontrolu jedů při léčbě jakéhokoli předávkování.
horní
Dávkování a správa
Doporučená dávka Provigilu je 200 mg podávaná jednou denně.
U pacientů s narkolepsií a OSAHS by se přípravek Provigil měl užívat jako jedna dávka ráno.
U pacientů se SWSD by se přípravek Provigil měl užívat přibližně 1 hodinu před zahájením pracovní směny.
Dávky až do 400 mg / den, podávané jako jedna dávka, byly dobře tolerovány, ale neexistuje žádný trvalý důkaz, že tato dávka poskytuje další přínos nad rámec dávky 200 mg (viz Klinická farmakologie a Klinické stezky).
Obecné úvahy
U souběžných léků, které jsou substráty pro CYP3A4, jako je triazolam a cyklosporin, je třeba zvážit úpravu dávky (viz Opatření, Lékové interakce).
Léky, které jsou do značné míry vylučovány metabolismem CYP2C19, jako je diazepam, propranolol, fenytoin (také prostřednictvím CYP2C9) nebo S-mefenytoin může být eliminován při současném podání s přípravkem Provigil a může vyžadovat snížení dávky a monitorování pro toxicitu.
U pacientů se závažným poškozením jater by měla být dávka Provigilu snížena na polovinu dávky doporučené pro pacienty s normální funkcí jater (viz CKlinická farmakologie a Opatření).
Pro určení bezpečnosti a účinnosti dávkování u pacientů se závažným poškozením ledvin nejsou k dispozici dostatečné informace (viz Klinická farmakologie a Opatření).
U starších pacientů může být v důsledku stárnutí eliminace Provigilu a jeho metabolitů snížena. Proto by mělo být zváženo použití nižších dávek v této populaci (viz Klinická farmakologie a Opatření).
horní
Jak dodáván
Provigil® (modafinil) Tablety
100 mg: Každá bílá, nepotahovaná tableta ve tvaru tobolky je na jedné straně vyražena „Provigil“ a na druhé straně „100 MG“.
NDC 63459-101-01 - Lahve po 100
200 mg: Každá bílá, potahovaná tableta ve tvaru tobolky je opatřena vyraženým „Provigil“ na jedné straně a „200 MG“ na druhé straně.
NDC 63459-201-01 - Lahve po 100
Uchovávejte při 20 ° - 25 ° C (68 ° - 77 ° F).
Vyrobeno pro:
Cephalon, Inc.
Frazer, PA 19355
U.S. RE37,516 / 4,927,855
© Cephalon, Inc., 2008. Všechna práva vyhrazena
PROV-011
Poslední aktualizace: 03/08
Informační list pacienta Provigil (modafinil) (v prosté angličtině)
Podrobné informace o Příznaky, příznaky, příčiny, léčba poruch spánku
Účelem informací v této monografii není pokrýt všechna možná použití, pokyny, preventivní opatření, lékové interakce nebo nepříznivé účinky. Tyto informace jsou zobecněné a neslouží jako zvláštní lékařská pomoc. Pokud máte dotazy týkající se léčivých přípravků, které užíváte, nebo byste chtěli získat více informací, obraťte se na svého lékaře, lékárníka nebo zdravotní sestru.
zpět k:
~ všechny články o poruchách spánku
